一项大型国际合作项目对38个癌症组织的2600多个肿瘤标本进行了全方位研究,对癌症的遗传基础提出了大量新见解。
自2001年首次完成人类基因组测序以来,对肿瘤进行全面基因组表征就成了癌症研究人员的主要目标。自那时起,测序技术和分析工具不断进步,推动了该领域的快速发展。《自然》2月6日发表的6篇论文中1-6,全基因组泛癌分析(Pan-Cancer Analysis of Whole Genomes,PCAWG)联盟呈现了迄今最全面也最庞大的癌症基因组荟萃分析。之前的分析主要聚焦癌症基因组中的蛋白质编码区域,而PCAWG分析的是整个基因组。每篇文章都侧重于癌症遗传学一个重要方面,综合起来看,这6篇文章对于全面掌握癌症的遗传复杂性具有重要意义。
在详细介绍每项研究之前,必须要指出的是,PCAWG背后依赖的是大量的数据和复杂的组织框架。整个项目依托四大洲的科学家组成的跨学科团队,一共涉及744个学术机构。为了在开展分布式研究的同时保护患者数据,科学家必须克服技术上、法律上和伦理上的重重阻碍。研究人员一共分为16个工作组,每个组专注于癌症基因组学的一个方面,例如,有的组评估突变重复率,有的组推断肿瘤的演化。
联盟一共对38种不同肿瘤进行了整合分析,对2658个全癌基因组进行了测序(图1),同时还对来自同一个体的相应非癌细胞样本进行了测序。此外,研究人员还分析了1188个肿瘤转录组——即肿瘤中RNA转录本的序列和丰富度。
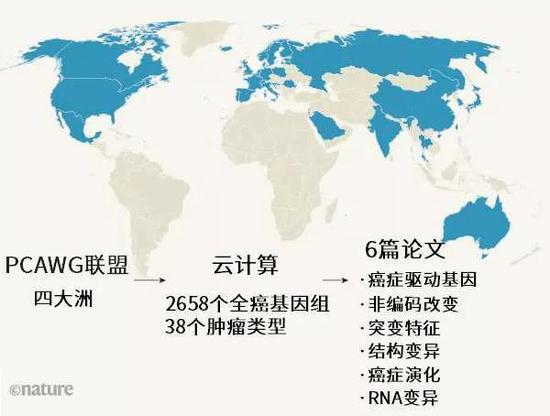
图1 | 全球性抗癌研究。全基因组泛癌分析(PCAWG)联盟由来自四大洲(蓝色)的癌症研究人员组成。联盟对38种肿瘤的2658个全癌基因组进行了测序和分析。这项工作涉及的大量数据需要动用复杂的云计算。联盟发表的6篇论文1-6分别侧重不同方面。(自然发表的作品对于发表地图中涉及的管辖权争议持中立态度。)
这些合作涉及广泛的质量控制,协调数据处理,以及对检测突变的不同计算流程进行大规模、系统性的实验验证。多个计算算法和流程必须同步使用和对比。这就需要几百TB的数据,这些数据分布在多个数据中心,可能需要百万小时的处理时间——而云计算极大缓解了这些问题。显然,PCAWG联盟的合作作为一个极佳的范例,证明了云计算在促进国际合作,推进数据密集型领域发展方面的关键作用。
第一篇论文1对PCAWG数据集的广度和深度进行了概述。联盟报告称,平均而言,每个癌症基因组携带4-5个驱动突变,这为癌细胞提供了选择优势。在被分析的肿瘤中,只有5%的肿瘤找不到任何驱动突变。相比之下,许多肿瘤都存在染色体重排(17.8%的肿瘤)和染色体碎裂(22.3%的肿瘤),这两个基因组灾难的典型表现会导致基因组发生重大的结构性变化。
其他5篇论文则从数据集的不同角度切入。在第2篇论文中,Rheinbay等人2鉴定了非编码DNA中的驱动基因。这项工作非常具有挑战性,因为检测非编码区中的突变比编码区中的突变要难得多,更不要说评估其重复率了。尽管如此,作者还是通过仔细建模、排除伪影,系统地鉴定出了非编码驱动突变。
作者的结果对之前报道的非编码驱动突变提出了质疑,如长链非编码RNA NEAT1和MALAT1。此外,作者也揭示了新的驱动突变,比如在关键肿瘤抑制基因TP53非编码区反复出现的一个突变。他们还发现端粒酶基因TERT的非编码区中存在频繁发生的突变,这些突变会导致端粒酶异常高表达(从而促进肿瘤细胞不受控地分裂)。这一发现印证了之前的一项泛癌研究的结果:进展期(转移性)肿瘤中的端粒酶突变率较高(12%)7。尽管这项研究并未直接排除其他非编码驱动突变的存在,但至少可以说明这类突变并不常见。
在第3和第4篇论文中,Alexandrov等人3和Li等人4主要关注被称为“特征”(signature)的基因组异常。不同的过程,如DNA修复机制存在缺陷或暴露于环境诱变剂,都会导致这种特征性的DNA变异。想要进一步提炼已知的突变特征,挖掘新的特征,就需要非常大的基因组数据集。值得称道的是,Alexandrov等人和Li等人共鉴定出97个特征。在这些新发现的特征中,不仅包含常规的单核苷酸特征,还包括与多核苷酸变异以及DNA小片段插入或缺失相关的特征。
不仅如此,Li和同事还是最早发现涉及结构变异(structural variant,SV)的可复制特征的研究团队之一,这里的结构变异是指大块基因组的重排。由于结构变异的多样性和复杂性,识别这类特征比识别突变特征要难得多。
通过对突变进行分组,研究人员一共鉴定出16个结构变异特征,揭示了缺失和相互倒位这两个结构变异之间的推断性机制联系等其他关系(最后一个特征涉及DNA片段方向的反转)。研究人员还对这16个特征在癌症中的作用提出了新见解。分析显示,特定DNA修复基因中的突变与一些癌症特征有关。比如,联盟发现,基因CDK12的突变与DNA串联重复有关;DNA修复酶MBD4的截短变异体会与涉及CpG位点这一DNA序列的突变特征同时出现。总而言之,这些新发现的特征为我们理解癌症发展机制以及诱变暴露在此过程中的作用奠定了基础。
1976年8,科学家首次提出,癌症的发展遵循一种演化的过程。自那以后,研究人员从随机突变和自然选择的角度来描述癌症演化的特征。如果癌细胞携带的突变具有高适应性,这类细胞就会迅速繁殖,成为细胞群中数量最多的细胞克隆。这种现象称为克隆清除(clonal sweep),会在肿瘤生长期间的反复发生。随着时间推移对同一个肿瘤的多个区域进行测序,是研究癌症演化的最有效方式;但研究人员也可以通过单次活检对演化过程进行重建——Gerstung等人5在第5篇论文中便采取了这一方法。
作者在这里引入了“分子时间”的概念,对克隆和亚克隆突变进行分类。他们推断,仅存在于一部分肿瘤细胞中的亚克隆突变,应该是在癌症演化的后期才出现的。对于所有肿瘤细胞中都存在的克隆突变,作者根据突变发生在克隆的拷贝数增益(一个基因或染色体区域的拷贝数增加)之前或之后,将克隆突变分为早期或晚期。研究人员将多种肿瘤的演化数据汇总,从而确定了一些常见的突变轨迹,比如APC-KRAS-TP539就是大肠癌中典型的突变发生顺序9。
Gerstung等人的研究发现,某种癌症中最常见的驱动突变,其出现时间往往也最早。同样,如果拷贝数增益在某种癌症中反复发生,其发生时间也较早。举例来说,在透明细胞肾癌中,5号染色体局部的拷贝数增益很常见,且多在肾癌发展早期就出现了。相反,全基因组复制在这类肾癌中的发生时间较晚。最后,研究人员发现,至少在40%的肿瘤中,突变特征会随时间变化。这些变化意味着,随着疾病进展,环境暴露的作用逐渐减弱,而DNA修复缺陷的出现频率和严重程度逐渐增加。总而言之,该小组的研究结果表明,驱动突变可以发生在癌症确诊的好几年前,这对疾病早发现以及生物标志物的开发都具有重要意义。
在最后一篇论文中,PCAWG转录组核心小组和他们的同事6分析了1188个肿瘤的转录组及全基因组测序数据,建立了DNA变异与RNA变异的功能联系。小组发现数百个单核苷酸DNA突变与附近基因的表达之间存在关联。然而,更大的拷贝数变异才是促使癌细胞中基因表达变化的主要因素。此外,突变还与转录本的结构变化有关,例如在非编码区(内含子)内形成新的蛋白质编码区(外显子)。
研究人员还描述了“桥式融合”(bridged fusion)的发生频率。桥式融合是指两个基因由于第三个DNA片段的插入而产生融合的现象。最终,在分析的1188个样本中,虽然87个在DNA水平上没有驱动变异,但每个样本中都发现了RNA水平的改变。总之,从这些结果可以看出,将RNA和DNA测序结果进行整合分析,对癌症研究具有重要作用10。
这6篇论文和其他期刊共同发表的相关论文(请参阅go.nature.com/3boajsm),可以看作是癌症和云基因组学领域的里程碑。通过分析推断,联盟成功将十年里以观察为主的癌症测序研究向前推进了一大步。值得注意的是,尽管与描述性研究相比,推理性分析能够增进我们对癌症的了解,但其结果的不确定性也更高。
PCAWG数据集的公开性和高质量,将会带来新一轮的生物学见解,推动方法学的发展。将其与其他功能性基因组数据集相整合,如探测基因组的3D架构,势必也会拓展我们对遗传学异常的原因及后果的认知。
当前研究最大的局限性在于,缺乏有关患者治疗及结局的临床数据。这些数据能帮助研究人员发现可预测临床结果的遗传学变化。幸运的是,一个名为国际癌症基因组联盟-加速基因组肿瘤学研究(ICGC-ARGO)的项目正在进行中,该项目将为10万多名癌症患者构建这样的资源库。
PCAWG汇集了千万科学家的力量,共同完成了这个使命。这些合作的长期影响不仅来自今天发表的研究结果,还将来自全球研究人员的通力协作,来自各成员之间的知识交流。
关键词: 癌症

















 营业执照公示信息
营业执照公示信息